As funçőes esquecidas do Oracle – Parte II
Olá, leitores, estamos de volta e continuaremos falando sobre algumas funçőes esquecidas do Oracle. Neste artigo falamos sobre Life Sciences e as funçőes que existem no banco de dados que săo usadas para dados biológicos. É isso mesmo! O Oracle (bem como o DB2) tem funçőes pré-definidas para serem usadas em dados de pesquisas com genomas, proteomas, vias metabólicas, etc.
As funçőes para alinhamento de seqüęncias podem ser encontradas dentro do banco dados e aplicadas diretamente sobre os dados armazenados no mesmo.
Bem, mas vamos dar uma refrescada na memória. O que é uma seqüęncia de dados de DNA? O que é um alinhamento de seqüęncias? Para que isto é importante? O que nós da computaçăo temos haver com essa coisa toda de biólogos e biomédicos? Vamos lá.
Uma seqüęncia de DNA ou seqüęncia genética é a representaçăo da estrutura primária de uma molécula de DNA. Essa representaçăo é feita através de letras, que representam os quatro tipos de nucleotídeos. As possíveis letras em uma seqüęncia de DNA săo: A, T, C e G que săo respectivamente Adenina, Timina, Citosina e Guanina. AAATCGCTTGG representa uma pequena seqüęncia de DNA; qualquer sucessăo de nucleotídeos maior que quatro já pode ser considerada uma seqüęncia.
Um alinhamento de seqüęncias é uma forma de descrever a relaçăo entre as cadeias de caracteres. Vejamos: se uma seqüęncia de DNA é representada por uma cadeia de caracteres, para compararmos duas seqüęncias nos iremos alinhá-las para descobrirmos onde podemos encontrar pontos similares. Esses pontos similares entre as seqüęncias podem indicar características iguais entre espécies diferentes e ajudam a traçar com maiores detalhes o processo evolutivo. Existem, basicamente, dois tipos de alinhamento: o global e o local. O alinhamento global consiste no alinhamento de no mínimo duas seqüęncias inteiras que nem sempre tęm o mesmo tamanho. Para suprir os espaços vagos entre uma seqüęncia e as outras săo levadas em conta algumas regras biológicas (que năo vamos entrar em detalhes) que permitem que as seqüęncias tenham, no final, tamanhos iguais. Já o alinhamento local é o alinhamento de apenas parte de uma seqüęncia com parte de outra e assim é possível comparar a similaridade entre determinadas funçőes descritas naquele pedaço da cadeia de DNA.
Vejam um pequeno exemplo:
Seqüęncia 1: CGCTATAT
Seqüęncia 2: TATACTA
O Alinhamento das duas ficaria desta forma:
CGCTATAT-T-
-----TATACTA
Acima podemos ver que as partes mais próximas estăo alinhadas. O coeficiente de similaridade entre as seqüęncias é obtido fazendo-se uma pontuaçăo (score) e, dada a pontuaçăo alcançada pelo alinhamento, é possível dizer o grau de similaridade entre elas. Năo vamos entrar no detalhe de como é feita esta pontuaçăo, pois envolvem alguns conceitos biológicos.
Estudos e trabalhos que envolvem esse tipo de análise representam uma grande evoluçăo em tratamentos médicos, produçăo de novos medicamentos, técnicas para entender o comportamento evolutivo de populaçőes, da humanidade, identificar fontes causadoras de doenças como o câncer, etc. E novamente eu pergunto. O que nós da computaçăo temos com isso? Sem a grande evoluçăo das máquinas e com isso, a possibilidade de implementarmos algoritmos com técnicas de inteligęncia artificial, servidores capazes de processar volumes extraordinários de dados, capacidade de armazenamento desses dados, etc. Sem tudo isso, os pesquisadores ainda estariam no início do processamento do genoma humano.
Uma das principais técnicas aplicadas em bioinformática é a de Mineraçăo de Dados, um dos focos desta coluna. As funçőes da Oracle que abordam o uso deste tipo de informaçăo fazem parte do pacote Oracle Data Mining.
Bom, depois desta rápida explicaçăo, vejamos na prática como usar a funçăo BLAST no Oracle.
BLAST é a abreviaçăo de Basic Local Alignment Search Tool. O BLAST foi um algoritmo desenvolvido em 1990 por Stephen Altschul para comparaçăo rápida de seqüęncias. Normalmente é usado para comparar seqüęncias de nucleotídeos ou de aminoácidos. Dentro do Oracle existem as seguintes variaçőes do BLAST:
· BLASTN: Compara uma seqüęncia de DNA informada pelo usuário com as seqüęncias armazenadas na base de dados e retorna as seqüęncias mais similares.
· BLASTP: Compara uma seqüęncia de proteínas informada pelo usuário com as seqüęncias armazenadas na base de dados e retorna as seqüęncias mais similares.
· BLASTX: Compara os produtos conceituais da traduçăo de uma seqüęncia de nucleotídeos com uma seqüęncia de proteína do banco de dados.
· TBLASTN: Compara uma seqüęncia de proteínas informada pelo usuário com seqüęncias de nucleotídeos no banco, fazendo a traduçăo de nucleotídeos para proteínas dinamicamente.
· TBLASTX: Compara as traduçőes das seqüęncias de nucleotídeos informadas pelo usuário com as seqüęncias de proteínas armazenadas na base.
Existem duas function tables para BLAST no Oracle:
MATCH, que retorna:
· q_seq_id: o identificador da seqüęncia.
· t_seq_id: identifica o id da seqüęncia usado em bancos específicos de seqüęncia, como GenBank.
· score: score do alinhamento.
· value: o valor esperado.
ALIGN, que retorna:
· q_seq_id: o identificador da seqüęncia.
· t_seq_id: identifica o id da seqüęncia usado em bancos específicos de seqüęncia, como GenBank.
· pct_identity: percentual de similaridade idęntica entre as seqüęncias consultadas e as armazenadas no banco.
· aligment_length: tamanho do alinhamento.
· mismatches: número de alinhamentos mal-sucedidos entre a seqüęncia informada e as armazenadas no banco.
· gap_openings: número de espaços inseridos para o alinhamento acontecer.
· gap_list: lista dos locais onde foram inseridos os espaços.
· q_start: posiçăo onde se inicia a inserçăo de espaço no alinhamento.
· q_end: posiçăo onde termina a inserçăo de espaço no alinhamento.
· s_start: posiçăo onde começa o alinhamento dentro da seqüęncia.
· s_end: posiçăo onde termina o alinhamento dentro da seqüęncia.
· expect: o valor esperado do alinhamento.
· score: score do alinhamento.
Nós vamos usar os dados abaixo para os nossos exemplos:
Tabela de Proteínas:
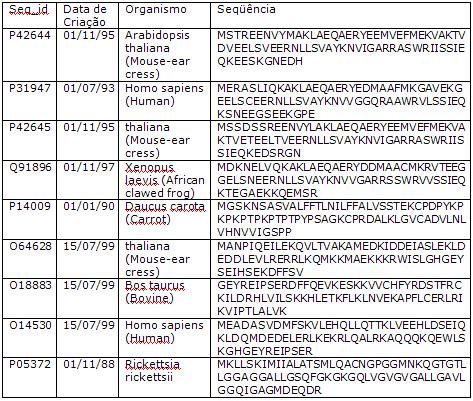
Tabela de Nucleotídeos – Organismo E. Coli:

Nosso primeiro exemplo é uma busca na seqüęncia de proteínas humanas nos dados do SwissProt (o SwissProt é um banco de dados de proteínas desenvolvido pelo Instituto de Bioinformática da Suíça). A seqüęncia que vamos informar também é uma parte de outra seqüęncia do SwissProt.
select T_SEQ_ID AS seq_id, score
from TABLE(
BLASTP_MATCH (
(select sequence from query_db),
CURSOR(SELECT seq_id, seq_data
FROM swissprot
WHERE organism = 'Homo sapiens (Human)'),
1,
-1,
0,
0,
'BLOSUM62',
10,
0,
0,
0,
0,
0)
)
order by score,seq_id;
SEQ_ID SCORE
---------- -----------
O14530 30
P31947 169
P31946 205
O score que tivemos como resposta representa o quăo próximo a seqüęncia em questăo está da seqüęncia informada. No nosso caso, a primeira seqüęncia se difere apenas 30 “pontos” da seqüęncia informada.
Nosso segundo exemplo utiliza o BLASTX, onde a seqüęncia que será inserida é de nucleotídeos, que será traduzida e comparada com dados de proteínas. Vamos usar as seqüęncias de nucleotídeos do organismo E. coli e a seqüęncia que iremos inserir é uma parte do SwissProt, banco de proteínas.
select *
from TABLE(
TBLAST_MATCH (
(select sequence from ecoli_query),
CURSOR(SELECT seq_id, seq_data FROM prot_db),
1,
-1,
'blastx',
1,
0,
0,
'BLOSUM62',
10,
0,
0,
0,
0,
0)
)
order by score,t_seq_id;
T_SEQ_ID SCORE
------------ ---------
103625 32
100368 33
103625 33
52624 33
54625 33
103625 36
132801 36
132801 38
132801 41
103625 44
132801 44
103625 45
12416 45
103625 49
Bem pessoal, se nós, além de usarmos estas funçőes específicas para dados biológicos pudermos juntar a isto outras características de BI, aumentamos as possibilidades de análise de dados. Por exemplo, poderíamos descobrir padrőes entre determinadas seqüęncias para criaçăo de novas drogas, apenas analisando as diferenças encontradas entre elas.
Eu espero que vocęs tenham gostado deste artigo e se tiverem mais interesse, mandem e-mail, postem comentários. Estou ŕ disposiçăo para esclarecer dúvidas.
Um abraço a todos e até a próxima.
Vander Emiro Muniz
vmuniz@triscal.com.br

